手性α-烷基吡咯烷酮广泛存在于具有生物活性的天然产物和药物分子中。目前,从易得的γ-内酰胺为原料高对映选择性合成α-单烷基的吡咯烷酮具有很大的挑战,因为存在潜在的过度烷基化,此外产物在碱性条件下容易消旋化。在药物化学领域,α-烷基的合成主要通过Evan的手性辅基完成不对称的α-烷基化,然后进行多种操作形成吡咯烷酮环。直到最近,不对称催化方法才应用于α-烷基化吡咯烷酮的合成。Zhang和Ding课题组报道环状芳基取代的烯酰胺的不对称氢化合成α-苄基取代的吡咯烷酮。Cramer课题组报道了镍催化的不对称氢甲酰化合成α-甲基取代的吡咯烷酮。

最近,华东理工大学得陈宜峰课题组,合成了8-喹啉咪唑啉配体Quinim,以简单易得得3-丁烯基氨基甲酰氯和烷基碘化物为原料,通过镍催化的分子内的还原偶联反应,高对映选择性地合成了一系列α-烷基取代的吡咯烷酮。
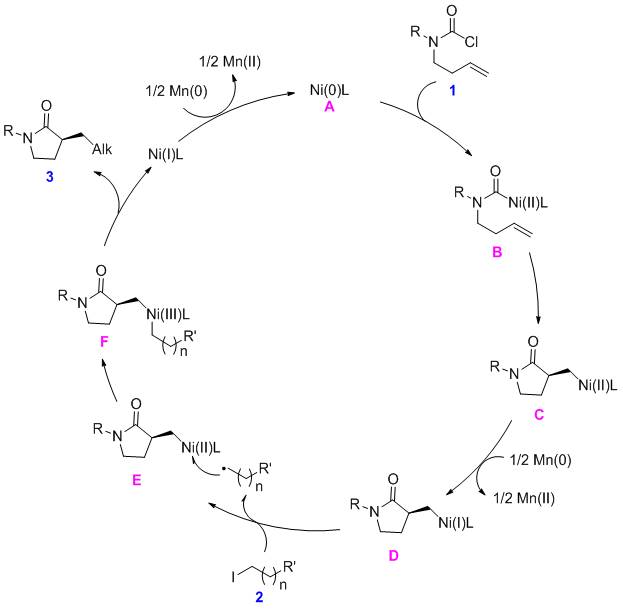
基于前人的报道和初步的理论研究,作者提出可能的反应机理:零价镍A和酰氯氧化加成得到二价镍物种B,然后分子内迁移插入,得到烷基镍中间体C,随着被锰还原产生中间体D。烷基碘代物与D通过单电子转移,形成中间体F,随后还原消除,得到产物3和一价镍物种,一价镍经锰还原,重生零价镍A完成催化循环。(J. Am. Chem. Soc. 2020, 142, 15654-15660)(推荐人:王新维,检查人:李翔)